Full articlegcms.jonasberdoz.dev →
I built a GC-MS processing pipeline professionally — taking raw chromatography data and turning it into identified molecules. It worked, but I kept thinking about a different approach. The kind of thought that doesn't go away.
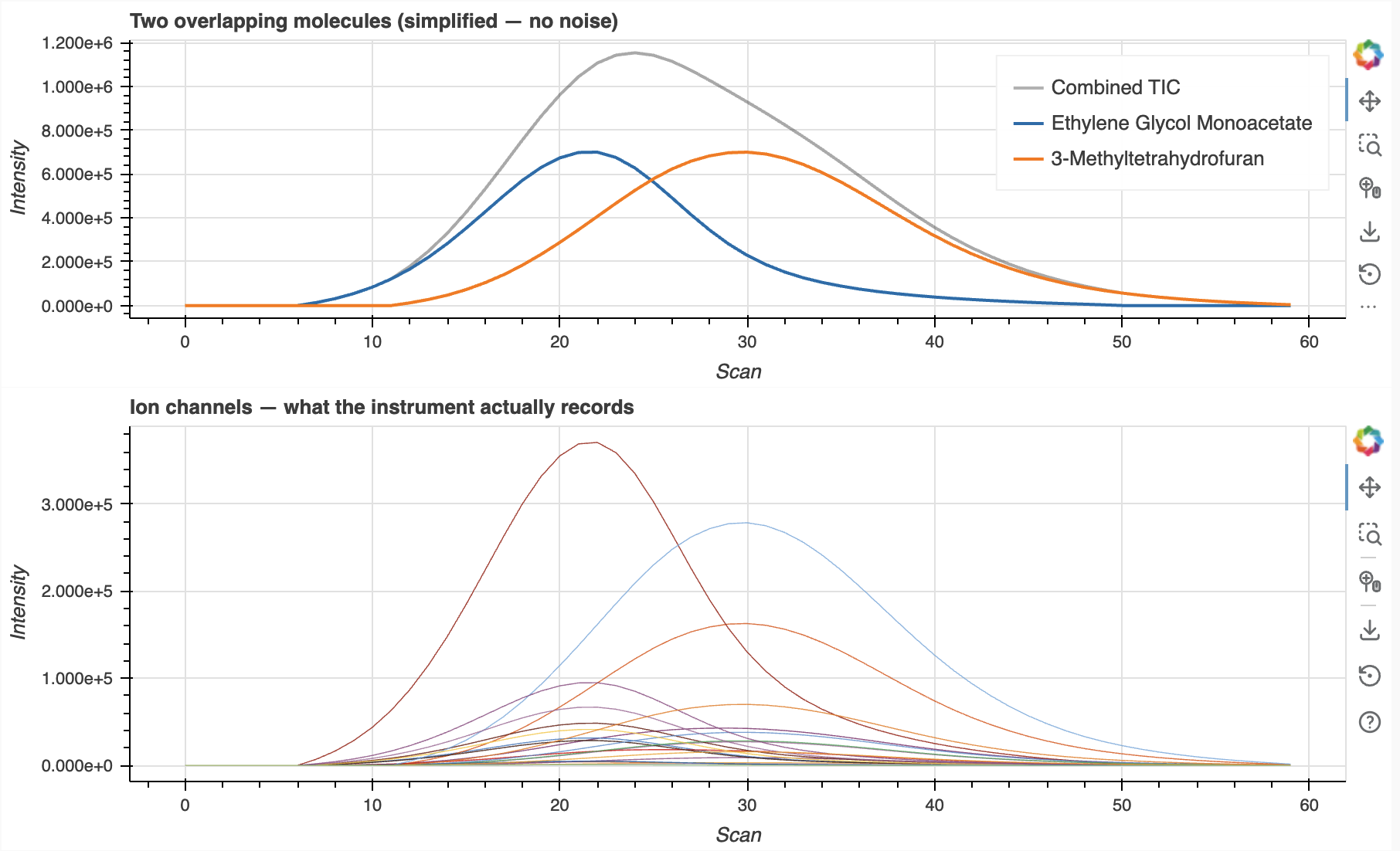
As I'm sometimes a bit obsessive, once an idea is stuck in my head, the only way to get rid of it is to build it. So I did, on my own time.
I didn't expect things to move as fast as they did. Working with Claude, the whole thing came together surprisingly quickly — preprocessing, peak detection, deconvolution, identification, ... It went so fast that building a small interactive blog around it was basically free.
It works great on synthetic data. On real data, not so much. But that's the honest part of the story, and I think it's worth telling.
Kudos to the researchers and open datasets behind all of this — the reference spectra and the deconvolution literature.